Our Research
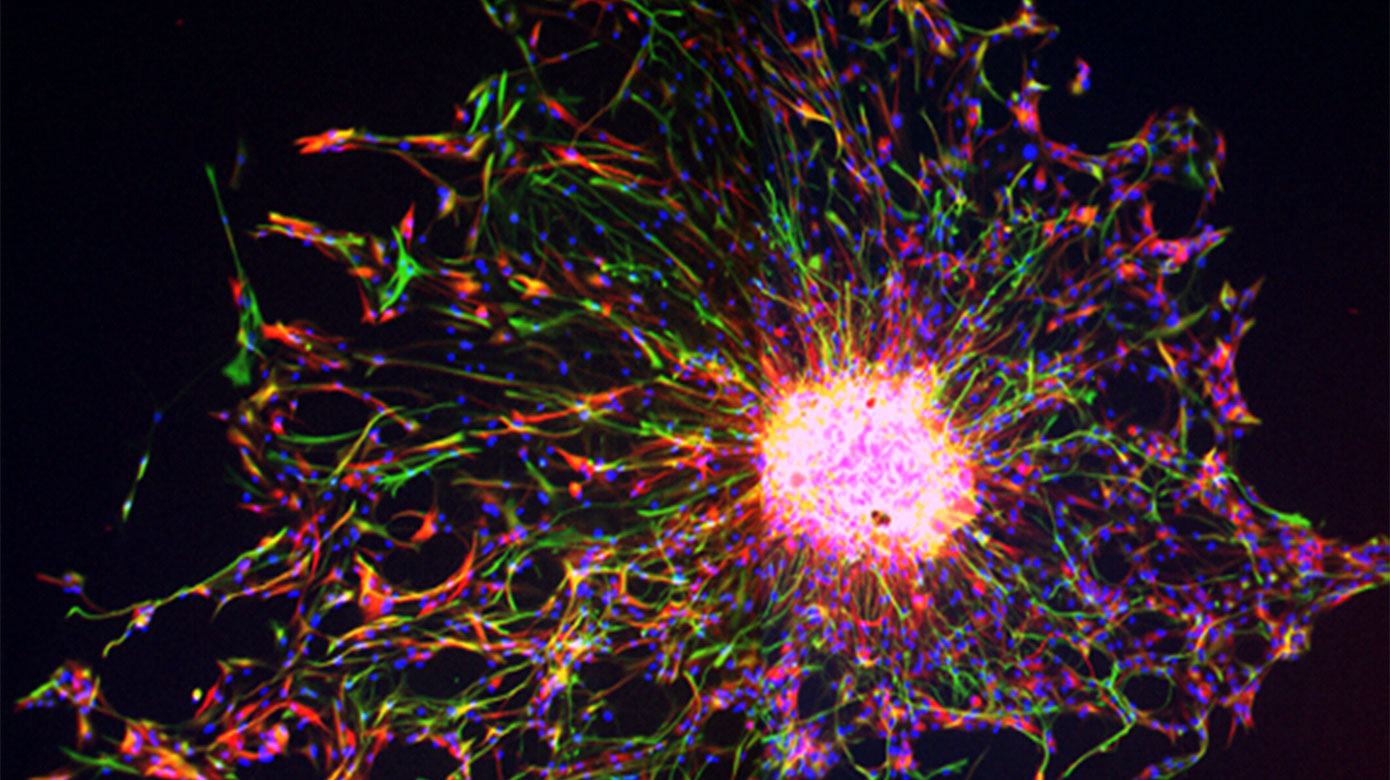
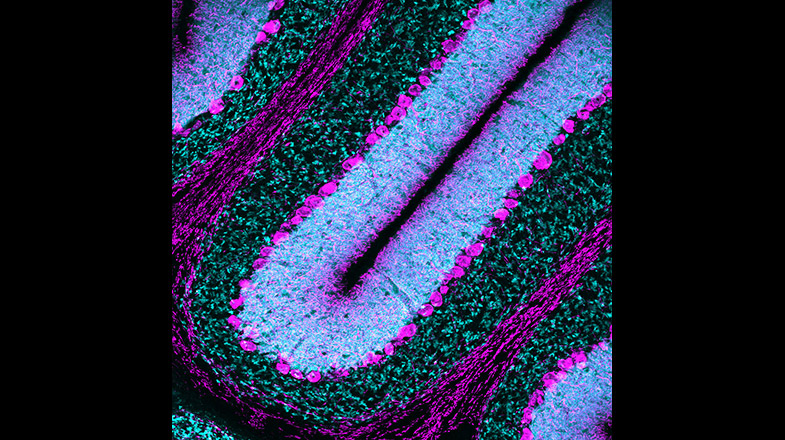
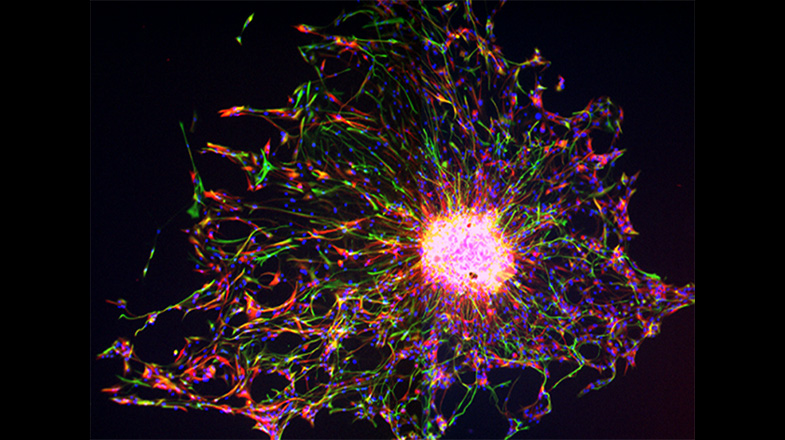
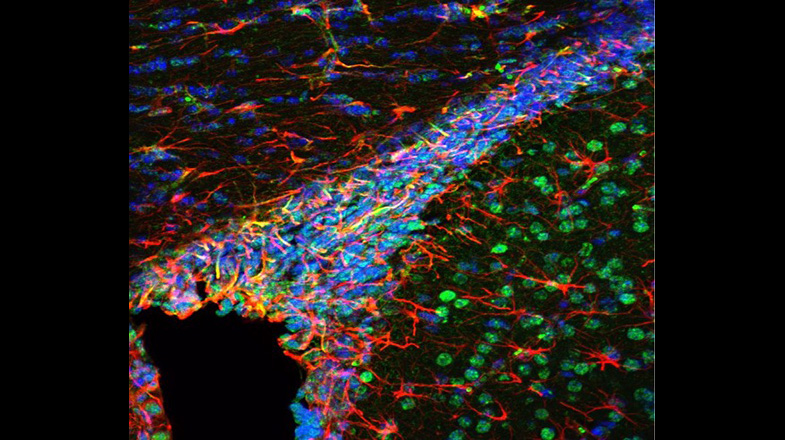
White Matter Development and Injury
A major cause of chronic disability in survivors of premature birth is diffuse white matter injury (DWMI) and hypomyelination. Altered development of the WM is directly associated with adverse outcomes, including cerebral palsy, cognitive delay and neurobehavioral problems. The cellular pathophysiology underlying DWMI and abnormal myelination is complex and not fully understood. WM glia, and particularly oligodendrocytes (OLs) and their progenitors (OPCs), are susceptible to injury that often occurs in premature birth. We have previously used an animal model of hypoxia (HX)-induced global WMI to demonstrate that OPCs display delayed maturation, which results in abnormal myelination and altered WM function. We have uncovered major aspects of the cellular dysmaturation pathology underlying HX-induced delayed myelination in corpus callosum, including enhanced OPC proliferation associated with decreased OL differentiation, and disrupted myelin ultrastructure. Recent analysis of OL development in corpus callosum (CC) demonstrates that: i) the prolonged proliferative state of OPCs and delayed OL differentiation in HX is a result of changes in HIF1α-dependent expression and activity of the histone deacetylases Sirt1 and Sirt2, respectively; ii) HX reduces synaptic glutamate (Glu) release from cortical pyramidal neurons on OPCs, which normally downregulates OPC proliferation, and iii) HX compromises axonal integrity and function in the subcortical white matter (SCWM), resulting in altered axon/myelin interactions. Based on these results, we are now testing the hypothesis that HX-induced protracted WM immaturity arises from intrinsic (Sirt1 and Sirt2) and extrinsic (synaptic) dysregulation of OPC proliferation and OL maturation, thus affecting axonal integrity and function.
Oligodendrocyte Development and Regeneration
In the postnatal and adult brain, OPCs arise from radial glial cells (RGCs) of the subventricular zone (SVZ), a major gliogenic and neurogenic region of the brain. OPC proliferate in the SVZ and migrate throughout the brain to gray and WM, where they mature into myelinating OLs. While some important signaling pathways have been characterized, much remains unknown about homeostatic regulation of OPC proliferation and maturation in the SVZ, both during normal development and after injury. Furthermore, although it is established that the proliferative response of endogenous OPCs to injury is crucial for expanding this progenitor pool and for regenerating a normal number of OLs, the endogenous molecular signals involved in the regulation of OPC proliferation in the SVZ are still largely undefined. We utilized our previously generated Endothelin-1 (ET-1) and ET-1 receptor (Ednr) mouse mutant lines and discovered that in the postnatal brain, RGC-derived ET-1 plays a novel and different role, i.e., regulates OPC proliferation. We are now testing the hypothesis that ET-1 signaling between RGCs and OPCs plays a crucial role in SVZ developmental homeostasis and regeneration.
Cerebellar Development, Function and Injury
Premature infants born earlier than 32 weeks with very low birth weight or VLBW (<1500 g) are highly vulnerable to developmental delay in sensory, cognitive and motor processing. Perinatal insults such as hypoxia (low O2) affect the developmental program of specific neuronal populations resulting in behavioral deficits. Between 24 to 32 gestational weeks, the cerebellar cortical surface area expands rapidly. Cerebellar circuitry, therefore, is prone to injury-related dysfunction during this time window. Clinical data indicates that VLBW infants are 40 to 100 times more likely to develop locomotor deficits than term infants. Using an established chronic perinatal hypoxia (Hx; 10.5% O2 from P3-P11) mouse model of neonatal brain injury, we previously identified a GABA-dependent developmental disruption of the cerebellar white matter. We also discovered a delay in the development of Purkinje cells (PCs), which constitute the sole output of the cerebellar cortex. In order to characterize potential behavioral and physiological consequences of PC developmental disruption, we are now using our model to investigate locomotor deficits and changes in PC electrophysiology, and how PC abnormalities are causally associated with locomotor impairment. We hypothesize that perinatal brain injury (Hx) leads to adaptive cerebellar behavioral deficits caused by abnormal Purkinje cell development and physiology.
Neurobiology and Neurophysiology of Down Syndrome
Down syndrome (DS) is the most commonly diagnosed chromosomal condition and the most common genetic cause of intellectual disability in the United States. DS affects a range of behavioral domains in children, including motor and cognitive function. Defective oligodendrocyte (OL) development and myelination have been identified in DS human brains and mouse models. This finding is consistent with studies demonstrating reduced white matter integrity in individuals with DS. The Gallo and Haydar Laboratories are collaborating to answer key questions regarding the timing and source of OL dysmaturation, and particularly whether cell-autonomous or non-cell-autonomous mechanisms lead to altered cellular differentiation and myelination. Our gene, protein and bioinformatics studies of human DS brain indicate impairments in the maturation of oligodendrocyte precursor cells (OPCs) into myelinating OLs (mOL). We have also shown that these same human brain gene and protein expression changes are found in the SCWM and cerebellum of the Ts65Dn mouse model of DS, and that they are correlated with significant in vivo abnormalities in OL differentiation and myelin production. Importantly, these cellular changes are accompanied by slower action potential conduction velocity between cerebral hemispheres, indicating that this newfound cellular deficit may play a large role in intellectual disability in DS. A detailed examination of the trisomic OLs and neurons is now warranted so that therapies can be designed to prevent the hypomyelination and its effects on motor and cognitive development in DS.
In a separate project, we are investigating locomotor dysfunction in DS. Clinical assessments indicate a range of locomotor deficits in DS, as well as slower adaptive control. Longitudinal data also indicates altered gait evolution from childhood to adulthood. Cerebellar pathology has been consistently observed in DS and is thought to contribute to dysfunction in locomotor and adaptive motor skills. Studies in animal models of DS have also indicated deficient cerebellar processing; however, the specific pathways underlying locomotor deficits and the cerebellar circuits that are disrupted in DS remain poorly understood. Defining specific abnormalities in motor behavior and identifying the brain regions and neurons which are functionally involved will provide the basis for developing potential therapies for treating motor problems in people with DS. The main goal of our studies is to identify specific alterations in the circuitry of the cerebellum that result in locomotor dysfunction in DS.